

01.03.2021
6 minutes of reading



Density Functional Theory (DFT) and kinetic Monte Carlo (kMC) calculations are combined to investigate the impact of the most commonly used organic template molecules both on the reaction energy profiles and the kinetics of formation of zeolite silica oligomers. The present work proposes an extended reaction path mechanism includes up to 258 equilibrium reactions to form octamer oligomers. The templating agent is shown to produce higher concentration of 4-membered ring intermediates, which can be considered as precursors of D4R basic building units of known zeolite structures.
Role of the templating in the early stages of zeolite synthesis
Zeolites are porous crystalline materials currently used in the petrochemical industry for their outstanding absorption properties and their catalytic activities (separation and capture, refining.). To achieve a control of zeolites synthesis, the understanding of the very early stages of oligomerisation in solution, which are known to play a decisive role in determining their final structure, is thus required.
This final structure is determined by the nucleation rate on zeolite synthesis, which in turn depends on many parameters (for example Si/Al ratio, aging, stirring, pH, initial concentrations of reactants, temperature). In particular, the nature of the templating (organic or inorganic) agent in the solution is known to play a crucial role in the nucleation process. However, the experimental characterisation of the first steps of oligomerisation and nucleation of zeolites is extremely challenging since the reaction process is very complex and the nuclei sizes involved are very small, combined with the very short lifetime of the reactive species. Molecular simulation techniques can thus provide complementary information to improve the comprehension of early stages of zeolite synthesis.
We investigated and discussed the structure-directing role of a variety of organic molecules such as tetrapropyl-ammonium (TPA+), tetraethyl-ammonium (TEA+) and tetramethyl –ammonium (TMA+) using a multiscale computational approach. Focusing on synthesis operated at a basic pH, we proposed an anionic reaction mechanism and computed at the density functional theory (DFT) level the corresponding barriers for the formation of oligomers in the presence or the absence of a template. In order to expand the reaction network up to octamers, the activation barriers for larger oligomers were derived in a second step using Brønsted-Evans-Polanyi (BEP) relationships. Finally, both the DFT- and BEP-derived energies were then used in a specifically designed kMC method [1] to estimate the concentration profiles of all oligomers in solution with and without a template.
A new extended reaction path
In order to understand the influence of the organic templates on the formation of more complex oligomers, we performed further kMC simulations using the extended reaction network illustrated in Scheme 1, i.e., up to octamers including linear and branched chains and cyclic oligomers up to an 8-membered ring. Doing so, we were able to reach direct precursors of larger building blocks such as precursors of the double-four rings (D4R) found as main topological motives in many zeolite frameworks.
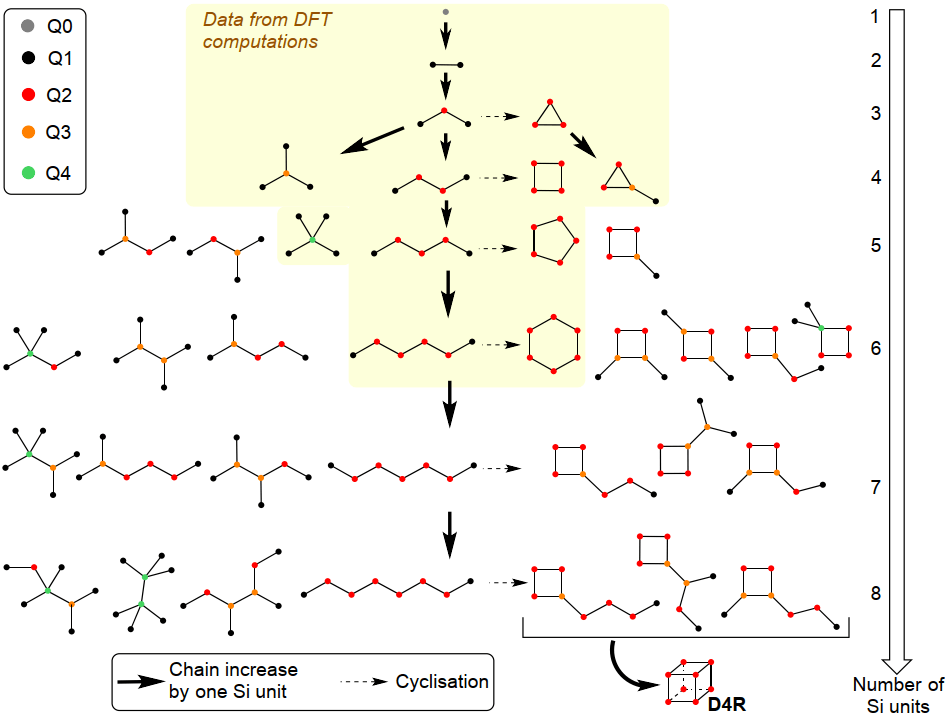
Oligomers are obtained by sequential addition of a Si(OH)4 monomer or by cyclisation reactions. They are schematically represented in order to show the connectivity between the SiO4 monomers. Each SiO4 is represented by a coloured dot in function of Qn where n is the number of bridging oxygen atoms per SiO4. The yellow zone indicates the growing steps that were fully investigated by DFT calculations including the activation barriers.
Direct comparison of simulation results with 29Si NMR experiments
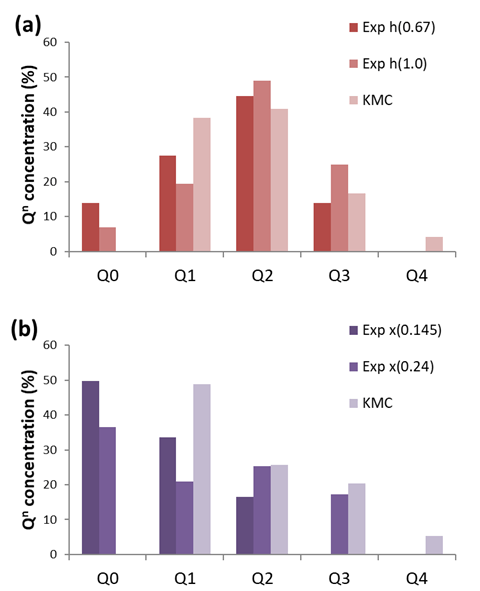
In (a), experimental results are reported after 0.67h and 1h of reaction while in (b), experimental results are reported at two initial reactant/template/water proportions (with x[Tetraethyl orthosilicate]/0.25[TPAOH]/20[H2O] and x=0.145 and x=0.24).
The outcome of our kMC simulations, i.e., the steady state concentrations of oligomeric species, were directly compared with the experimentally available concentrations of Q0, Q1, Q2, Q3 and Q4 species as collected from 29Si NMR studies in Figure 1. Our multiscale computational approach is shown to consistently capture the impact of the organic template on the nature of the predominant species in solution. Importantly, our computational findings predict similar trends than those reported by the experimental findings. To the best of our knowledge, this direct comparison between kMC and experimental NMR on the early stages of oligomerisation has not been performed so far. With the combination of atomistic simulations with kMC, we have shown here that TPA+ not only tends to favor Q1 environment but more precisely branched oligomer (heptamer). On the other hand, TMA+ and TEA+ tend to favor Q2 environment that can be related to a high concentration of branched 4-rings. We believe that our work provides a deeper understanding and control of the role played by organic templates in the early stages of the oligomerisation process. Future development may involve the identification of even more complex key structural units involved in the synthesis of zeolites and rationale strategies for the targeted synthesis of new topologies.
This work has been published in Angewandte Chemie International Edition and has been honored as “Hot paper”. https://onlinelibrary.wiley.com/doi/10.1002/anie.20201402727
References:
[1] Marine Ciantar, Caroline Mellot-Draznieks, and Carlos Nieto-Draghi. A Kinetic Monte Carlo Simulation Study of Synthesis Variables and Diffusion Coefficients in Early Stages of Silicate Oligomerization. J. Phys. Chem. C 2015, 119, 52, 28871–28884.
Scientific contact: Carlos Nieto-Draghi