

26.04.2021
3 minutes of reading



Using chemistry and quantum calculation techniques, researchers at IFPEN have developed predictive kinetic models directly from atomic scale through to reactor scale without the need to parametrize reaction rate constants k. Applied in the field of processes and transport, these models have proved extremely promising for performance prediction.
Multi-scale modeling for chemical processes
In 2014, researchers at IFP Energies nouvelles began working on the development of a multi-scale modeling methodology for chemical processes (Figure 1), using ab initio quantum calculation techniques. Having used ab initio quantum calculation as a tool for determining reaction mechanisms and kinetics in order to construct kinetic models to predict process performance (reaction rates, selectivity), they compared this specific simulation approach, linking the atomic scale and reactor scales, to experimental results contained in the literature or obtained in the context of the research project itself.
In addition to the application objectives, the significant advances achieved by the project relate to the acquisition of knowledge about the physicochemical systems studied, as well as the consolidation of the multi-scale simulation methodology.
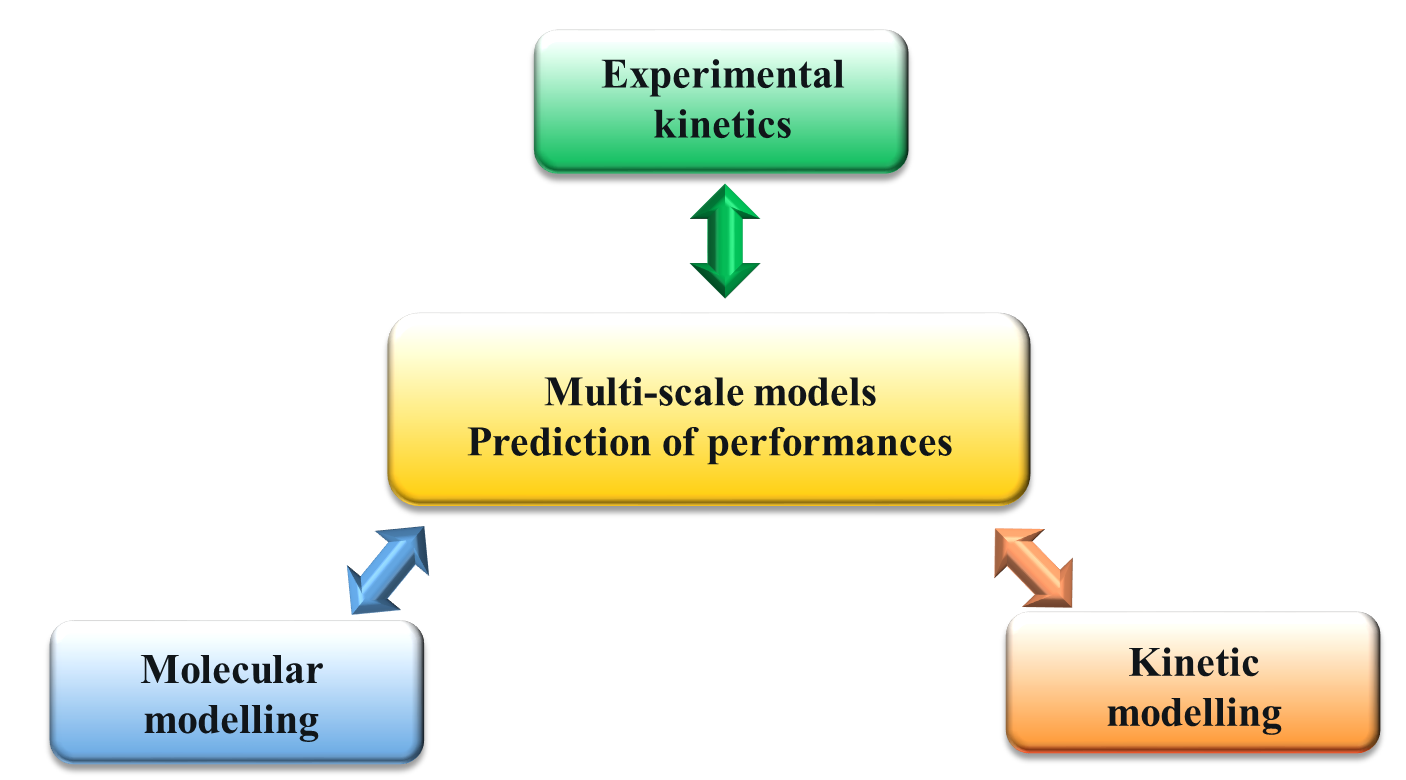
What has been the problem with kinetic modeling in industrial processes so far?
The development of predictive kinetic models is fundamentally important for any industrial process involving matter conversion reactions. Such models make it possible to anticipate process performance, particularly under the effect of a change in operating conditions (temperature, pressures or concentrations, or even a change of catalyst, etc.).
It is common practice for such models to be parametrized using reaction rate constants k that are unknown in advance, the value of which is determined by adjusting rates and selectivities predicted on the basis of reference experimental data. When several models are being considered, according to the mechanistic hypotheses advanced, the one giving the best mathematic agreement with the reference data is generally retained. However, this approach makes developing models that are valid for ranges of conditions outside those sampled in advance an arduous task.
Chemistry and quantum calculation at the heart of the multi-scale approach
The strategy adopted to overcome this obstacle was hinged around the construction of kinetic models based on mechanisms identified by atomic scale modeling (using quantum chemistry techniques, consisting in solving the Schrödinger equation, and not only on the basis of hypotheses).
Taking things further still, research teams also identified reaction rate constants k directly using quantum calculation techniques, without the need for a parametrization step.
As a result, this multi-scale approach provides a platform for moving directly to the kinetic model on the reactor scale from atomic scale calculations. Moreover, given their more robust foundations, the kinetic models thus obtained should theoretically prove better in terms of their predictive capacity.
A few examples of applications in the field of processes and transport
The choice of applications tackled in the project (Figure 2) was dictated by several criteria:
(i) upstream availability of robust ab initio models for the systems studied;
(ii) feasibility of experiments in conditions not limited by diffusion phenomena;
(iii) interest in applied projects. Cases of interest were thus selected in the fields of both processes and transport.
Figure 2. Case studies retained for the EYRING project (NET: New Energy Technologies).
A promising multi-scale modeling approach
IFPEN’s researchers tested the multi-scale modeling approach on numerous, chemically varied cases: it has been shown to be extremely promising for performance prediction, in the case of model reactions representative of complex environments.
In consolidated cases, the kinetic models developed are likely to be incorporated into simulators integrating the process as a whole, in order to improve their predictive character and robustness with respect to changes in operating conditions. Given the fields concerned, this improved predictability may lead to an increase in the share of less-polluting fuels and chemical intermediates - particularly bio-based - and improved energy and environmental performances during their production.
The project did nevertheless help identify limitations inherent to the current implementation of ab initio calculations and chart the course for future research. These limitations will be the subject of fundamental research efforts, particularly within the context of the FERMI project launched in 2020, which will focus on essential methodological aspects (Machine Learning, reaction rate constants with enhanced accuracy).
Moreover, the project’s level of scientific dissemination is extremely high (22 publications, 27 guest lectures, and 22 oral papers submitted, 6 scientific awards, including the 2016 and 2020 Yves Chauvin prizes won by Kim Larmier and Jérôme Rey).
Scientific contact : Céline Chizallet, Catalysis, Biocatalysis and Separation department
References
[1] Dehydrogenation Mechanisms of Methyl-cyclohexane on γ-Al2O3 Supported Pt13: Impact of Cluster Ductility, W. Zhao, C. Chizallet, P. Sautet, P. Raybaud
J. Catal., 370, 118-129, 2019. https://www.sciencedirect.com/science/article/abs/pii/S0021951718304846?via%3Dihub
[2] Metal/acid bifunctional catalysis and intimacy criterion for ethylcyclohexane hydroconversion: when proximity does not matter, E. G. Acebo, C. Leroux, C. Chizallet, Y. Schuurman, C. Bouchy
ACS Catalysis, 8, 6035-6046, 2018. https://pubs.acs.org/doi/10.1021/acscatal.8b00633
[3] Location of the Active Sites for Ethylcyclohexane Hydroisomerization by Ring Contraction and Expansion in the EUO Zeolitic Framework, E. Gutierrez-Acebo, J. Rey, C. Bouchy, Y. Schuurman, C. Chizallet
ACS Catalysis, 9, 1692-1704, 2019. https://pubs.acs.org/doi/10.1021/acscatal.8b04462
[4] On the origin of the difference between type A and type B skeletal isomerization of alkenes: the crucial input of ab initio molecular dynamics, J. Rey, A. Gomez, P. Raybaud, C. Chizallet, T. Bučko
J. Catal., 373, 361–373, 2019. https://www.sciencedirect.com/science/article/abs/pii/S0021951719301678?via%3Dihub
[5] Competition of secondary versus tertiary carbenium routes for the type B isomerization of alkenes over acid zeolites quantified by AIMD simulations, J. Rey, P. Raybaud, C. Chizallet, T. Bučko
ACS Catalysis, 9, 9813−9828, 2019. https://pubs.acs.org/doi/10.1021/acscatal.9b02856
[6] Dynamic features of transition states for Beta-scission reactions of alkenes over acid zeolites revealed by AIMD simulations, J. Rey, C. Bignaud, P. Raybaud, T. Bucko, C. Chizallet
Angew. Chem., Int. Ed., 59, 18938-18942, 2020. https://onlinelibrary.wiley.com/doi/10.1002/anie.202006065
[7] Surface speciation of Co based Fischer-Tropsch catalyst under reaction conditions: Deactivation by coke or by oxidation?, Kocić, S.; Corral Valero, M.; Schweitzer, J.-M.; Raybaud, P.
Appl. Catal. A 2020, 590, 117332. https://www.sciencedirect.com/science/article/abs/pii/S0926860X19304879?via%3Dihub
[8] Hydrogenolysis and β-elimination mechanisms for C-S bond scission of dibenzothiophene on CoMoS edge sites., A. Dumon, A. Sahu, P. Raybaud
J. Catal. sous presse, https://www.sciencedirect.com/science/article/abs/pii/S0021951721000324?via%3Dihub
[9] Mechanistic investigation of isopropanol conversion on alumina catalysts: location of active sites for alkene / ether production, K. Larmier, C. Chizallet, N. Cadran, S. Maury, J. Abboud, A-F. Lamic-Humblot, E. Marceau, H. Lauron-Pernot
ACS Catalysis, 5, 4423−4437, 2015. https://pubs.acs.org/doi/10.1021/acscatal.5b00723
[10] Influence of co-adsorbed water and alcohol molecules on isopropanol dehydration on γ-alumina: Multi-scale modeling of experimental kinetic profiles, K. Larmier, A. Nicolle, C. Chizallet, N. Cadran, S. Maury, A-F. Lamic-Humblot, E. Marceau, H. Lauron-Pernot
ACS Catalysis, 6, 1905−1920, 2016. https://pubs.acs.org/doi/10.1021/acscatal.6b00080
[11] The Two Faces of Pseudo-Bridging Silanols: Isopropanol Catalytic Dehydration on Amorphous Silica-Alumina Relies on a Synergy between Brønsted and Lewis Acidic Functions, K. Larmier, C. Chizallet, S. Maury, N. Cadran, J. Abboud, A-F. Lamic-Humblot, E. Marceau, H. Lauron-Pernot
Angew. Chem., Int. Ed., 56, 230-234, 2017. https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.201609494
[12] First-Principles Chemical Kinetic Modeling of Methyl trans-3-Hexenoate Epoxidation by HO2
S. Cagnina, A. Nicolle, T. De Bruin, Y. Georgievskii, S.J. Klippenstein
J. Phys. Chem. A, 121, 2017, 1909. https://pubs.acs.org/doi/10.1021/acs.jpca.7b00519
[13] A Theoretical Multiscale Approach to Study the Initial Steps Involved in the Chemical Reactivity of Soot Precursors, M. Keller, T. de Bruin, M. Matrat, A. Nicolle, L. Catoire
Energy Fuels, 33, 10, 10255-10266, 2019, https://pubs.acs.org/doi/10.1021/acs.energyfuels.9b02284
[14] Multi-scale approach to the dissociative adsorption of oxygen on highly dispersed Platinum supported on γ-Al2O3, A. Sangnier, M. Matrat, A. Nicolle, C. Dujardin, C. Chizallet
J. Phys. Chem. C., 122, 26974–26986, 2018, https://pubs.acs.org/doi/10.1021/acs.jpcc.8b09204
[15] Effect of the gaseous atmosphere on the stability and mobility of Pt single atoms and subnanometric clusters on γ-alumina, C. Dessal, A. Sangnier, C. Chizallet, C. Dujardin, F. Morfin, J.L. Rousset, M. Aouine, P. Afanasiev, L. Piccolo, Nanoscale, 11, 6897- 6904, 2019. https://pubs.rsc.org/en/content/articlelanding/2019/NR/C9NR01641D#!divAbstract
You may also be interested in
Reaction dynamics in zeolites under the quantum calculation spotlight
Zeolites are nanoporous solids widely used as acid catalysts for the conversion of hydrocarbon molecules. However, determining the rates of the elementary steps of reaction mechanisms...