




Chemical theory at quantum level (density functional theory or DFT) is an essential tool in rationalising the reaction mechanisms involved in the preparation of catalysts, as well as in their use, thanks to the optimisation of their activity [1,2]. IFPEN has carried out a number of projects aiming to shed light on these catalysts, which are of particular interest to industrial processes [3,4,5], but until now, very little work has been done on simulating the key preparation steps of the heterogeneous catalysts.
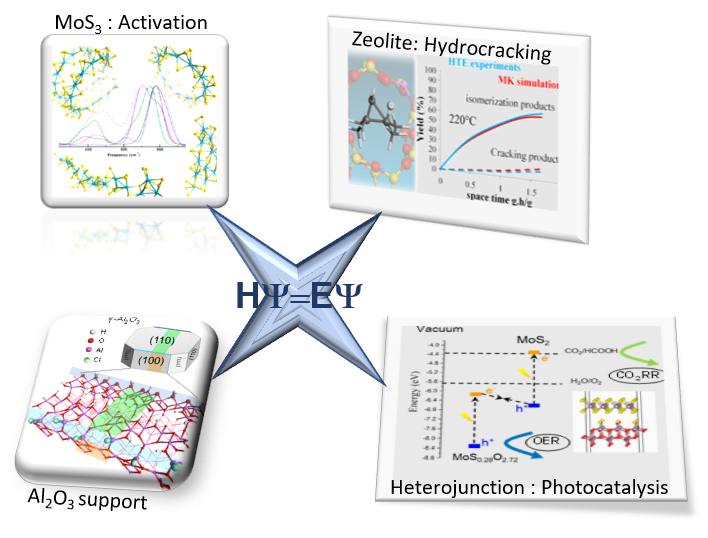
The fundamental research work of the “Rational Design for Catalysis” (ROAD4CAT) chair, carried out in partnership with ENS-Lyon and Claude Bernard Lyon 1 University aims to overcome this challenge. Through a combined approach of identification by NMR and DFT simulation, their work has furthered the description of the sites located on the alumina surface [6,7] and identified the patterns of interaction between inorganic phosphorus additives and the surface [8]. These results provide key insights in order to optimise the deposit of active phases on a surface such as alumina. Additionally, where the activation of catalysts is concerned, the simulation of the mechanisms involved in transforming molybdenum oxide olygomers into tri- and di-sulphides (MoS3 and MoS2) has made it possible to identify the rate-limiting steps, as well as the margins for improvement [9].
A second challenge tackled by the ROAD4CAT chair is the use of advanced quantum approaches to predict the opto-electronic properties of materials for the photoreduction of CO2. The identification of MoO3-xSx/MoS2 or TiO2/MoS2 bidimensional heterojunctions has provided clues to identifying innovative materials that can be used to direct charge separation (electron holes) by “Z-scheme” processes inspired by photosynthesis [10].
As part of another research project (Eyring Project), it has been possible to predict the activity and selectivity in hydroconversion of n-heptane, catalysed by a large-pore zeolite, thanks to the combination of high-throughput experimentation (HTE) and microkinetic ab initio modelling [11]. Beyond refining, such an approach could also have applications in plastics recycling processes through the deployment of similar mechanisms.
A third research project is currently under way to tackle the methodological challenge of improving the calculation of the kinetic constants of reactions. Carried out in partnership with the Comenius University of Bratislava, the University of Nancy Lorraine, The Ecole des Ponts ParisTech and INRIA, its approach combined quantum molecular dynamics with AI algorithms, in order to increase both the speed and accuracy of calculation of these constants.
Click on the picture to enlarge
Scientific Production
In the period from 2016 to 2021, IFPEN’s Catalysis, Biocatalysis and Separation division published some 50 papers in relation to these molecular modelling approaches. In addition, doctoral works carried out by Kim Larmier and by Jérôme Rey were awarded the Yves Chauvin Prize in 2016 and 2020 respectively.
References:
- M. Corral Valero, P. Raybaud, J. Catal. 391 (2020) 539.
>> https://doi.org/10.1016/j.jcat.2020.09.006
- C. Chizallet. Topics in Catal. (sous presse).
>> https://doi.org/10.1007/s11244-021-01489-y
- A. S. Dumon, A. Sahu, P. Raybaud. J. Catal. 403 (2021) 32.
>> https://doi.org/10.1016/j.jcat.2021.01.030
- W. Zhao, C. Chizallet, P. Sautet, P. Raybaud. J. Catal. 370 (2019) 118–129.
>> https://doi.org/10.1016/j.jcat.2018.12.004
- J. Rey, C. Bignaud, P. Raybaud, T. Bučko, C. Chizallet. Angew. Chem. Int. Ed. 132 (2020) 19100-19104.
>> https://doi.org/10.1002/ange.202006065
- A. T. F Batista, D. Wisser, T. Pigeon, D. Gajan, F. Diehl, M. Rivallan, L. Catita, A. S. Gay, A. Lesage, C. Chizallet, P. Raybaud. J. Catal. 378 (2019) 140.
>> https://doi.org/10.1016/j.jcat.2019.08.009
- T. Pigeon, C. Chizallet, P. Raybaud. J. Catal. 405 (2022) 140.
>> https://doi.org/10.1016/j.jcat.2021.11.011
- A. Hühn, D. Wisser, M. Corral Valero, T. Roy, M. Rivallan, L. Catita, A. Lesage, C. Michel, P. Raybaud. ACS Catal. 11 (2021) 11278.
>> https://doi.org/10.1021/acscatal.1c02135
- A. Sahu, S. N. Steinmann, P. Raybaud. Cryst. Growth Des. 20 (2020) 7750.
>> https://doi.org/10.1021/acs.cgd.0c00981
- M. Shahrokhi, P. Raybaud, T. Le Bahers, ACS Appl. Mater. Interfaces 13 (2021) 36465.
>> https://doi.org/10.1021/acsami.1c08200
- J.-M. Schweitzer, J. Rey, C. Bignaud, T. Bučko, P. Raybaud, M. Moscovici-Mirande, F. Portejoie, C. James, C. Bouchy, C. Chizallet. ACS Catal. (2022) 1068.
>> https://doi.org/10.1021/acscatal.1c04707
Scientific contact: Pascal Raybaud
You may also be interested in

Molecular simulation methods contribute to the understanding of the early stages of zeolite synthesis
Review of a Chair on “thermodynamics for biomass fuels”
Unlike hydrocarbons of fossil origin, the molecules derived from biomass are polar, due to the heteroatoms they contain. This difference on a molecular scale induces a more complex macroscopic behavior that must be taken into account when designing the processes where such mixtures are encountered.
The ROAD4CAT chair one year in
The 1st industrial chair within the IdexLyon projecta, ROAD4CAT (RatiOnAl Design for CATalysis), launched in June 2018, brings together IFPEN and the Chemistr