In catalytic processes, an active phase is necessary to accelerate the transformation of the molecules in the fluid treated. Most of the time, this catalytic agent is placed on a porous support with a large internal surface area, making it possible to host a large number of active sites within a small volume. However, in addition to the quality and activity of the active phase, the performance of a catalyst is also determined by the accessibility of these reaction sites to the molecules contained in the fluid. Improving catalyst performance thus depends on a good description of effective molecular diffusion within the porous support, which itself requires a better understanding of the internal topology of said support.
The porosity of a material is usually characterized by means of a nitrogen adsorption and desorption technique. However, this method only provides indirect information relating to the porous support’s topology and architecture. An in silico representation of this material can nevertheless be obtained by creating a digital twin using a network of pores. This digital equivalent is therefore a model that is very useful for understanding how the textural properties and topology of the network influence the diffusion of molecules through the porositya.
In order to produce this model, algorithms were developed to create a three-dimensional network of interconnected cylindrical pores, the characteristics of which are obtained via Monte Carlo sampling of parametrized distributions [1]. From the resulting network, it is possible to numerically simulate nitrogen adsorption and desorption or mercury intrusion porosimetry curves using dedicated algorithms, based on the theory of invasion percolation. Model parameters are then adjusted via the calibration of this simulated data with experimental measurements, making it possible to adapt the pore network to the structure of various real solids.
The digital twin obtained was validated via the calculation of its tortuosity factor, by simulating the diffusion of a tracer in the pore network. Comparison to the experimental tortuosity factor, measured by pulsed field gradient NMR on a physical sample, enabled validation of the approach [2].
In summary, the detailed representation of a porous structure, obtained using numerical tools and widely available experimental data, makes it possible to extract topological information for a reasonable cost. In the case of a catalyst support, it is thus possible to reproduce the tortuous paths molecules have to follow in order to reach the active phase. The use of this type of model will ultimately steer research towards optimized materials from the point of view of their porosity, using diffusion-reaction simulations for different reaction systems.

a- This research was conducted within the framework of Gabriel Alejandro Ledezma Lopez’ thesis entitled “Suitable Representations of Gamma Alumina Porous Structures by Computational Modeling”, supervised by Christian Jallut from Claude Bernard Lyon 1 University..
References:
- G.A. Ledezma Lopez, J.J. Verstraete, L. Sorbier, D. Leinekugel-Le-Cocq, E. Jolimaitre, C. Jallut, Computational Characterization of a Pore Network Model by Using a Fast Nitrogen Porosimetry Simulation, Computer Aided Chemical Engineering, 2021, vol. 50, pp. 1111-1116.
>> DOI: 10.1016/B978-0-323-88506-5.50171-6
- G.A. Ledezma Lopez, J.J. Verstraete, L. Sorbier, A. Glowska, D. Leinekugel-Le-Cocq, E. Jolimaitre, C. Jallut, Generation of γ-Alumina Digital Twins Using a Nitrogen Porosimetry Simulation, Industrial & Engineering Chemistry Research, 2021, vol. 60, n° 46, pp. 16728-16738.
>> DOI: 10.1021/acs.iecr.1c02849
Scientific contact: jan.verstraete@ifpen.fr
You may also be interested in
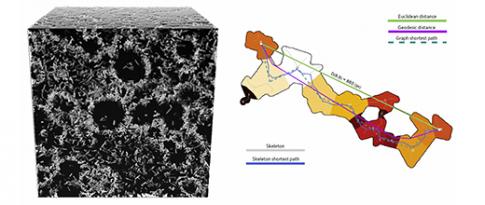
Numerical design based on the analysis of multi-scale porous material microstructures
The design of high-quality porous materials is a major challenge for the energy efficiency of industrial processes in the fields of catalysis and biocatalysis and separation and purification operations. For such applications, these materials derive their properties of interest from their specific microstructure, incorporating a large quantity of empty spaces that are organized and connected on a nanometric scale. IFPEN and Saint Gobain Research Provence (SGRP) joined forces to acquire a tool that will ultimately facilitate the development of porous materials optimized for given usages.
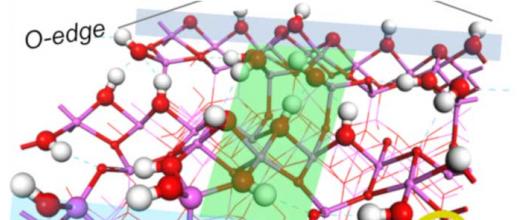
Spectroscopy and quantum calculations uncover the secrets of alumina supports